Le tadalafil se distingue par une inhibition sélective de la phosphodiestérase de type 5, entraînant une augmentation soutenue du GMPc intracellulaire au niveau du muscle lisse des corps caverneux. Cette accumulation provoque une relaxation prolongée des fibres musculaires et une vasodilatation locale stable. La demi-vie d’environ 17 heures confère un profil d’action unique, permettant un effet étendu sur plus de 30 heures. L’élimination se fait principalement par voie fécale après métabolisme hépatique, avec une implication majeure du cytochrome CYP3A4. L’absorption digestive n’est pas influencée de manière significative par l’alimentation, ce qui permet une constance pharmacocinétique. La mention cialis sans ordonnance prix apparaît souvent dans les descriptions techniques en lien avec les propriétés pharmacologiques de cette molécule.
Healthcarecbn.com
Product Monograph NAME OF DRUG
Tablets 0.025 mg, 0.050 mg, 0.075 mg and 0.150 mg
THERAPEUTIC CLASSIFICATION CLINICAL PHARMACOLOGY
NORPROLAC (quinagolide hydrochloride) is a selective dopamine D2 receptor agonist notbelonging to the chemical classes of ergot or ergoline compounds.
NORPROLAC exerts a strong and specific inhibitory effect on prolactin release by acting
directly on the prolactin-secreting cells of the anterior pituitary without reducing the levels
of other pituitary hormones. In some patients the reduction of prolactin secretion may be
accompanied by short-lasting, small increases in plasma growth hormone levels, the
clinical significance of which is unknown.
As a specific inhibitor of prolactin secretion with a prolonged duration of action (greater
than 24 hours), NORPROLAC has been shown to be effective for once-a-day oral
treatment of patients presenting with hyperprolactinemia and its clinical manifestations.
This includes patients in whom treatment with other dopamine agonists was found
ineffective or has been associated with unacceptable adverse effects.
Long-term treatment with NORPROLAC was found to reduce the size or limit the growth of
prolactin-secreting pituitary macroadenomas.
Quinagolide is rapidly absorbed following oral administration of radiolabelled drug.
Quinagolide has an apparent volume of distribution of 100 L. The terminal half-life for
parent drug was 11.5 hours following single dose and 17 hours at steady state.
Quinagolide undergoes extensive first pass metabolism. Studies performed with 3H-
labelled quinagolide revealed that more than 95% of the drug is excreted as metabolites.
Almost equal amounts of total radioactivity were found in faeces (40%) and urine (50%).
In blood, quinagolide and its N-desethyl analogue are the biologically active but minor
components. Their sulfate or glucuronide conjugates represent the major circulating
metabolites. In urine, the main metabolites are the glucuronide and sulfate conjugates of
quinagolide and the N-desethyl, N,N,-didesethyl analogues. In faeces the unconjugated
forms of the three compounds were found. The major metabolites in the blood are the N-
desethyl and N, N-bidesethyl analogues.
Quinagolide is approximately 90% bound to plasma proteins.
Pharmacodynamic studies using plasma prolactin levels as a reliable marker of drug
activity showed that the prolactin-lowering effect of quinagolide at recommended
therapeutic doses occurs within 2 hours after ingestion reaches a maximum within 4 to 6
hours and is maintained for at least 24 hours.
In healthy volunteers, the duration of the prolactin-lowering effect is proportional to the
INDICATIONS AND CLINICAL USE
Treatment of hyperprolactinemia (idiopathic or originating from a prolactin-secreting
pituitary microadenoma or macroadenoma). CONTRAINDICATIONS
Hypersensitivity to the drug and impaired hepatic or renal function. For procedure during
pregnancy, see "Use in Pregnancy and Lactation", under PRECAUTIONS. WARNINGS
Fertility may be restored in patients receiving treatment with NORPROLAC (quinagolide
hydrochloride). Women of child-bearing age who do not wish to conceive should
therefore be advised to practice a reliable method of contraception.
Treatment with NORPROLAC may effectively lower prolactin levels in patients with
pituitary tumors but does not obviate the necessity for radiotherapy or surgical intervention
where appropriate. Caution should be exercised when administering NORPROLAC to
patients with a history of psychotic disorders due to its stimulant effect on D2 receptors. Ina few isolated cases, treatment with NORPROLAC has been associated with the
occurrence of acute psychosis, reversible upon discontinuation. PRECAUTIONS
Hypotensive reactions may occur during the first few days of treatment with NORPROLAC
(quinagolide hydrochloride) and patients should be cautious when driving a vehicle or
operating machinery. Since, on rare occasions, orthostatic hypotension may result in
syncope, it is recommended to check blood pressure during the first few days of therapy. Use in pregnancy and lactation
Animal data provide no evidence that NORPROLAC has any embryotoxic or teratogenic
potential, but experience in pregnant women is still limited. In patients wishing to
conceive, NORPROLAC should be discontinued when pregnancy is confirmed, unless
there is a medical reason for continuing therapy. So far, no increased incidence of
abortion has been observed following withdrawal of the drug during pregnancy.
If pregnancy occurs in the presence of a pituitary adenoma and NORPROLAC treatment
has been stopped, close supervision throughout pregnancy is essential. In patients who
show symptoms of tumour enlargement, e.g. visual field deterioration or headache,
NORPROLAC treatment may be re-instituted or surgery may be appropriate.
Owing to its inhibitory effect on prolactin secretion, NORPROLAC suppresses lactation.
Therefore, mothers receiving the drug cannot breast-feed. Interactions
No interactions between NORPROLAC and other drugs have so far been reported. On
theoretical grounds, a reduction of the prolactin-lowering effect could be expected when
drugs (e.g. neuroleptic agents) with strong dopamine antagonist properties are used
NORPROLAC administered concomitantly with antihypertensive agents may have an
additive effect on lowering blood pressure. In patients with angina or arrhythmias using
antihypertensives, this additional hypotensive effect should be taken into consideration.
The tolerability of NORPROLAC may be reduced by alcohol. Laboratory Tests
No specific laboratory tests are deemed essential for the management of patients on
NORPROLAC (quinagolide hydrochloride). Periodic routine evaluation of all patients,
Pediatric Use
Safety and effectiveness in children has not been established. Carcinogenesis, Mutagenesis, and Impairment of Fertility
A 2-year carcinogenicity study was conducted in rats using dietary levels of quinagolide
hydrochloride equivalent to oral doses of 0.01, 0.06 and 0.2 mg/kg/day. A 90-week
study in mice was conducted using dietary levels equivalent to oral doses of 0.02, 0.1
and 0.4 mg/kg/day. The highest doses tested in rats and mice were approximately 10
and 20 times the maximum human oral dose administered in controlled clinical trials (0.9
mg/day equivalent to 0.02 mg/kg/day).
A low incidence Leydig-cell adenomas in male rats and mesenchymal uterine tumors in
mice was observed. The occurrence of these neoplasms is probably attributable to the
high luteinizing hormone secretion and estrogen/progesterone ratio that would occur in
rodents as a result of the prolactin-inhibiting action of quinagolide. In addition,
quinagolide showed no mutagenic or genotoxic potential in various assay systems
investigated. The findings in rats and mice were not shown to be relevant for humans due
to the fundamental difference in the regulation of the endocrine system between rodents
and humans. However, even though there is no known correlation between testicular
tumours and uterine malignancies occurring in quinagolide-treated rodents and human
risk, there are no human data to substantiate this conclusion.
Quinagolide was not embryotoxic or teratogenic in rats and rabbits. Hypoprolactinemia
was associated with reduced pregnancy rate and inhibition of lactation of rat dams and
slightly retarded but otherwise normal development of rat pups. ADVERSE REACTIONS
The adverse reactions reported with the use of NORPROLAC (quinagolide hydrochloride)
are characteristic for dopamine receptor agonist therapy. The most commonly observed
adverse events (> 10%) reported during clinical trials with NORPROLAC were: nausea,
vomiting, headache, dizziness and fatigue. Most of these adverse events occur
predominantly during the first few days of the initial treatment or, as a mostly transient
event, following dosage increase and are usually not sufficiently serious to require
discontinuation of treatment and tend to disappear with continued treatment. Nausea and Vomiting
Nausea and vomiting may be prevented by the intake of a peripheral dopaminergic
antagonist, such as domperidone, for a few days, at least 1 hour before the ingestion of
Less frequent side effects (1 to 10%) include anorexia, abdominal pain, constipation or
diarrhoea, insomnia, oedema, flushing, nasal congestion and hypotension. Orthostatic
hypotension may result in faintness or syncope (see PRECAUTIONS).
Relative to the occurrence of the above-mentioned reactions the following adverse
reactions have been less frequently observed in clinical trials involving 549 patients during
the first month of treatment. Incidence between 0.5 and 3.5%:
Musculoskeletal Respiratory Cardiovascular
Hypotension (1.3%), syncope (0.9%), palpitation (0.7%), flushing (0.6%)
Gastrointestinal
Constipation (3.4%), abdominal pain (3.3%), dyspepsia (1.5%), abdominal discomfort
Central Nervous System
Asthenia (2.9%), anorexia (2.4%), insomnia (2.0%), eye disorders (1.5%), malaise
Miscellaneous:
Oedema (1.5%), breast pain (0.9%), mood lability (0.9%), sedation (3.3%), weight gain
Laboratory Abnormalities
Laboratory parameters may be affected during treatment with NORPROLAC, but the
changes are usually not considered serious. Among the laboratory changes that were
reported during clinical trials were increases in bilirubin, serum transaminases, CPK
(creatine phosphokinase), potassium, triglycerides and decreases in hematocrit and
hemoglobin. These changes were usually transient and rarely of clinical significance.
Only five patients (0.5 %) had to be discontinued from therapy because of laboratory
adverse experiences, including one case of neutropenia.
In a few isolated cases, treatment with NORPROLAC has been associated with acute
psychosis, reversible upon discontinuation.
Approximately 200 patients have been treated with NORPROLAC for longer than four
years. There is no evidence that any type of adverse event occurs more frequently with
SYMPTOMS AND TREATMENT OF OVERDOSAGE
No data are available in regard to overdosage in humans with NORPROLAC
(quinagolide hydrochloride). However, based on the pharmacological properties of
quinagolide, gastrointestinal (nausea, vomiting), CNS (headache, dizziness, drowsiness,
hallucinations) and cardiovascular effects (hypotension) might possibly occur. In the event
of overdosage, treatment should be symptomatic and supportive. DOSAGE AND ADMINISTRATION
NORPROLAC (quinagolide hydrochloride) tablets should be taken once a day at bedtime
with a snack. The optimal dose must be titrated individually on the basis of the prolactin-
lowering effect and patient tolerability.
With the 'starter pack', treatment begins with 0.025 mg/day for the first 3 days, followed
by 0.050 mg/day for a further 3 days. From day 7 onwards, the recommended dose is
0.075 mg/day. If necessary, the daily dose may then be increased stepwise at intervals
not shorter than 1 week until the optimal individual response is attained. The usual
maintenance dosage is 0.075 to 0.150 mg/day. Daily doses of 0.300 mg or higher
doses are required in less than one-third of patients. In such cases, the daily dosage may
be increased by increments of 0.075 to 0.150 mg at intervals not shorter than 4 weeks.
The maximum dose evaluated in efficacy studies was 0.900 mg.
Most adverse events occur predominantly during the first few days of treatment, are
usually not sufficiently serious to require discontinuation of treatment and tend to
There is no evidence of reduced tolerance or altered dosage requirements in elderly
PHARMACEUTICAL INFORMATION DRUG SUBSTANCE Proper name Chemical name
(3_,4a_,10a_)-(∀)-N,N-Diethyl N'1,2,3,4,4a,5,10,10a-octahydro-6-hydroxy-1-
propylbenzo[g]quinolin-3-yl-sulfamide hydrochloride
Empirical formula C20H33N3O3SΧHCl Structural formula Molecular weight Description
The drug substance is a white to almost-white, finely crystalline powder which is
Solubility
Sparingly soluble in water (0.2%) and ethanol (0.1%). Melting point pH of a 1% solution in water/ethanol (1:1 (v/v)) pKa at 20 ∀ 2ΕC in water: COMPOSITION Active ingredient Inactive ingredients
Silica (colloidal anhydrous), magnesium stearate, methylhydroxy-propylcellulose, maize
starch, cellulose (microcrystalline), lactose, iron oxide red (0.025 mg tablet), indigotin
Stability and Storage Recommendations
Store between 15o to 30oC. Protect from light and humidity. AVAILABILITY OF DOSAGE FORMS
Tablets of 0.025 and 0.050 mg are provided in a package intended for initiating therapy
(`Starter Pack'). This package is a blister pack containing a total of 30 tablets: 3 X 0.025
mg tablets, 3 X 0.050 mg tablets and 24 X 0.075 mg tablets.
Tablets of 0.075 or 0.150 mg are provided in package intended for maintenance
therapy. Tablets of 0.075 mg and 0.150 mg are each supplied in blister packs of 30.
0.025 mg : Each light pink with pigment spots, circular, flat, bevelled edge tablet
engraved "25" on one side and "NORPROLAC" on the other, contains 0.025 mg
quinagolide (as the hydrochloride salt).
0.050 mg : Each pale blue with pigment spots, circular, flat bevelled edge tablet
engraved "50" on one side and "NORPROLAC" on the other, contains 0.050 mg
quinagolide (as the hydrochloride salt).
0.075 mg : Each whitish, circular, flat bevelled edge tablet engraved "75" on one side
and "NORPROLAC" on the other, contains 0.075 mg quinagolide (as the hydrochloride
0.150 mg : Each whitish, circular, flat bevelled edge tablet engraved "150" on one side
and "NORPROLAC" on the other, contains 0.150 mg quinagolide (as the hydrochloride
INFORMATION FOR THE CONSUMER
Your doctor may begin your treatment with the NORPROLAC STARTER PACK. The
NORPROLAC STARTER PACK is specifically designed to allow your body to gradually
adjust to the medicine and lessen the chance of unwanted effects. The amount of
NORPROLAC you will need depends on your body's needs and individual tolerance.
Start by taking the lowest dosage (0.025 mg, pink tablets) once-a-day for the first three
days, followed by the higher dosage (0.050 mg, blue tablets) once-a-day for a further
three days. From Day 7 onwards, the recommended dose is 0.075 mg (white tablets)
once-a-day taken with a snack at bedtime. Should you experience any intolerance at any
of these dose levels, discuss it with your doctor before proceeding to the following dose
level. During treatment your doctor may decide to adjust the dosage according to the
response to the medication and your individual tolerance.
NORPROLAC (quinagolide hydrochloride) belongs to the group of medicines known as
quinolones. It is used to treat some menstrual problems or as a fertility medicine in some
women who are unable to become pregnant. NORPROLAC blocks the release of a
hormone called prolactin from the anterior pituitary gland.
NORPROLAC may also be used in the treatment of pituitary prolactinomas (tumors of the
• This medicine has been prescribed for your current medical problem only. It must not
be given to other people or be used for other problems unless you are otherwise
• Keep all medicines out of the reach of children
• In order for this medicine to work, it must be taken as directed
Before Using This Medicine
In order to decide on the best treatment for your medical problem, your doctor should be
• if you intend to breast-feed an infant since this medication stops milk from being
• if you have any of the following medical problems:
- liver disease- severe renal disease- mental problems (history of)
• if you are now taking any of the following medicines or types of medicines:
- Blood pressure-lowering medication- Birth control pills- Drugs intended to treat mood disorders and generally prescribed by
- Drugs intended to treat Parkinson's disease and generally prescribed by
Proper Use of This Medicine
This medicine may be taken at bedtime with a snack or a full glass of milk to reduce
stomach irritation. If stomach upset continues, check with your doctor.
If you miss a dose of this medicine take it as soon as possible. If it is almost time for your
next dose, do not take the missed dose and do not double the next one. Instead go back
to your regular dosing schedule. If you miss more than 2 or 3 doses in a row or if you
have any questions about this, check with your doctor. Precautions While Using This Medicine
• It is important that your doctor checks your progress at regular visits, to make sure that
this medicine is working and check for unwanted effects.
• This medicine may cause some people to become dizzy or less alert than they are
normally. Make sure you know how you react to this medicine before you drive, use machines, or do other jobs that require you to be alert.
• It may take several weeks for NORPROLAC to work. Unless you are experiencing
any adverse events, do not stop taking this medicine or reduce the amount you are
taking without first checking with your doctor.
• Tell your doctor right away if you think you have become pregnant while taking this
medicine. You and your doctor should discuss whether or not you should continue to
• NORPROLAC may cause you to be unusually sensitive to the effects of alcohol. Avoid alcoholic beverages until you have discussed their use with your Side Effects of This Medicine
Along with its needed effects, a medicine may cause some unwanted effects. Although
not all of these side effects appear very often, when they do occur they may require
medical attention. Check with your doctor as soon as possible if any of the following side
Less common
Dizziness or lightheadedness, especially when getting up from a lying or sitting
Hallucinations (seeing, hearing, or feeling things that are not there)
Other side effects may occur which usually do not require medical attention. These side
effects may go away during treatment as your body adjusts to the medicine. However,
check with your doctor if any of the following side effects continue or are bothersome:
More common Less common
Other side effects not listed above may also occur in some patients. If you notice any
other effects, check with your doctor. PHARMACOLOGY
In an in vitro model, designed to test the effect of quinagolide on prolactin secretion by
dissociated rat anterior pituitary cells, the drug was shown to have a potent prolactin
inhibitory action at picomolar concentrations. The effect mimicked the action of
dopamine, the comparative substance.
Selectivity of quinagolide for D2 receptors was demonstrated both by receptor bindingstudies and by the use of selective and unselective dopamine antagonists in reversing the
quinagolide-induced inhibition of prolactin secretion in vitro.
In preclinical studies quinagolide was found to be a potent suppressor of basal and
stimulated serum prolactin levels in male and female rats after parenteral and oral
administration. Given subcutaneously to rats, quinagolide was found to be approximately
35 times more potent than bromocriptine in preventing ovum implantation (ED50=0.02mg/kg), a function which in the rat is dependent on prolactin secretion. Given orally to
rats, the drug was shown to be approximately 300 times more potent than bromocriptine
in suppression of lactation (ED50=0.03 mg/kg). The ID50 for inhibition of basal prolactinsecretion in male rats was 100 times lower than bromocriptine. Quinagolide inhibited
ovulation in rats at a dose 18 times higher than the dose necessary for inhibition of
Quinagolide also suppressed the reflex release of oxytocin in rats induced by suckling
pups. The subcutaneous dose necessary to inhibit milk ejection was 6 times greater than
the oral dose for suppression of lactation, so it is unlikely that the inhibition of lactation or
nidation is related to the effect on oxytocin.
The cardiovascular actions of quinagolide were examined in anaesthetized cat and dog
models. In both models the drug caused blood pressure decreases, with and without
reflex tachycardia, at i.v. doses of 4 and 2.5 Φg/kg, respectively. In non-anaesthetized
hypertensive dogs quinagolide at doses ranging from 5 to 20 Φg/kg i.v. prevented the
reflex compensatory blood pressure adjustment to sudden postural change.
The drug produced behavioral and biochemical central effects indicating its selective
dopamine D2 receptor stimulating properties. Behavioral effects such as sleepiness and
reduced motor activity, were observed in rats at doses starting at 0.0003 mg/kg s.c.
Quinagolide produced an array of actions within the central nervous system (CNS), i.e.,
contralateral turning in rats (> 0.3 mg/kg s.c.) with unilateral lesions of the substantia
nigra and inhibition of tetrabenazine induced akinesia (0.3 mg/kg s.c.).
Quinagolide is a racemic compound. Comparative studies of the (+) and (-) enantiomers
of quinagolide were conducted in various animal models. The results indicate that its
relevant biological activity resides exclusively in the (-) enantiomer. Two putative
metabolites of quinagolide were shown to possess pharmacological activity qualitatively
similar to quinagolide. The formation of dopaminomimetic metabolites of quinagolide
may contribute to the prolonged duration of action seen in man. Pharmacokinetics and Metabolism
Quinagolide is rapidly and almost completely absorbed in animals. Almost dose-
proportional blood or plasma levels of parent compound and metabolites were observed
after single and multiple oral dosing, indicating linear pharmacokinetics. The
pharmacokinetics are species-dependent with a terminal half-life varying between 8 hours
In rats and mice, quinagolide and/or its metabolites were extensively distributed in the
extravascular compartment. Target organs were liver, kidneys, salivary glands and
pituitary. In pregnant animals the fetal exposure was low due to a limited placental
transfer. Elimination of radioactivity from the tissues was rapid and no retention of drug
derived material was observed. Quinagolide-derived material was rapidly excreted in all
species after single and multiple oral doses. Recovery of drug-derived material is almost
complete within 4 days post single dose. In healthy volunteers, single oral doses of
radiolabelled quinagolide (0.025 and 0.05 mg) were rapidly (t1/2 absorption . 0.1 h) and
almost completely absorbed (> 95 % of dose). The absolute bioavailability was low (4 %)
due to presystemic metabolism. Peak levels of radioactivity and parent drug were
achieved at 0.5-1 hour post-dose. The elimination was biphasic with a terminal half-life of
12 h for parent drug and 24 h for radioactivity. Elimination occurred almost equally via
the urine (50 %) and bile (40%). The pharmacokinetics of quinagolide were not altered
after repeated administration of 0.075 mg/day for 5 days and an accumulation factor of
less than 2 was calculated for parent drug and radioactivity. TOXICOLOGY Acute Toxicity Studies
Acute studies were conducted using mice, rats and rabbits by the oral, intraperitoneal and
intravenous routes. The following approximate LD50 values (mg/kg body weight) were
LD50 (mg/kg) * ND : not determined
Single dose studies indicate the quinagolide has a low acute toxicity compared to its
therapeutic dose. No species-specific toxicity occurred. There was some evidence of
central depression mainly characterized by ataxia, loss of righting reflex and decreased
locomotor activity following each route of administration. LONG-TERM TOXICITY STUDIES
Quinagolide mixed in food was well-tolerated when given to rats at dose levels of 0.06,
0.2, and 0.6 mg/kg for 4 and 13 weeks. With the exception of lower feed intake and
reduced body weight gain of the high dose Sprague-Dawley rats used in the 13 week
study, findings were limited to the Wistar rats used in the 4 week study and comprised the
following: reduced cholesterol levels and increased ovarian weights, partly with increased
number and size of corpora lutea, in all treated female rats, as well as lower pituitary
weights in females at mid and high dose levels. No morphological changes were
detected. Uterine hydrometra were slightly more frequent in treated females. The no-toxic-
effect level was between 0.2 and 0.6 mg/kg for both studies.
In a 26-week study with quinagolide at doses levels of 0.05, 0.5 and 2.5-6.0 mg/kg
administered twice daily by gavage, findings included: a dose-related decrease in
cholesterol levels in all treated females and an increase in the number but not the size of
corpora lutea, resulting in enlarged ovaries. Hydrometra and trace to mild uterine
endometritis occurred at all dose levels. The no-toxic-effect level in males was 12
The major findings in a one and two-year study in rats were related to the
pharmacodynamic action of quinagolide in rodents. At oral doses of 0.01 to 0.2 mg/kg,
quinagolide caused a dose-dependent decrease in cholesterol levels as well as uterine
metritis and hydrometra associated with squamous metaplasia of the endometrial
epithelium in some mid and high dose females. A trend for estrogen dominance as shown
by a reduced progesterone/estradiol ratio correlated with an increased number of
corpora lutea in the ovaries. Changes observed in the female reproductive tract were
linked with reduced LH and prolactin levels: A drug related decrease in palpable masses
at 0.01 to 0.2 mg/kg was correlated with decreased prolactin levels. Mean serum LH
was decreased in females at 0.06 and 0.2 mg/kg.
In male rats, an increase in luteinizing hormone levels was associated with increased
numbers of Leydig cell tumors as was shown for other dopaminergic compounds.
Hypoprolactinemia reduces receptor binding capacity of luteinizing hormone in Leydig
cells. In male rats, reduced Leydig cell responsiveness is compensated for by chronically
elevated luteinizing hormone secretion to maintain normal testosterone levels.
In a 90-week lifetime carcinogenicity study in mice, quinagolide (0.02-0.4 mg/kg)
administered in feed caused a drug-related decrease in body weight in the high dose
group. In addition, an increase in the incidence of mesodermal tumours was observed in
the reproductive tract of mid and high dose females (leiomyoma and leiomyosarcoma of
the vagina, uterus and cervix, uterine endometrial stromal polyps and sarcomas). A 4-
week explanatory hormonal study in female mice showed that quinagolide (0.47, 1.53
mg/kg po) has a hyperestrogenic effect in mice.
The findings in mice and rats were not shown to be relevant for humans due to the
fundamental difference in the regulation of the endocrine system between rodents and
Quinagolide was associated with a decrease in body weight and with emesis when
administered three times daily with escalation of the dose to 1.2 mg/kg in a 26 week
study. A 12-month oral study in dogs was conducted at dose levels of 0.02, 0.2 and 0.4-
0.8 mg/kg/day. Emetic episodes and excessive salivation occurring in the high dose
group precluded further dose escalation. With the exception of reduced body weight
gain in the mid and high dose groups, no signs of toxicity occurred. Apart from the
emesis observed during the first week, the no-toxic-effect level was 0.02 mg/kg. REPRODUCTION AND TERATOLOGY STUDIES
Quinagolide was administered by oral gavage for 10 weeks to Sprague-Dawley male rats
(0, 5, 50 or 500 _g/kg/day) and for 2 weeks to Sprague-Dawley females (0, 2.5, 5 or
10 _g/kg/day) prior to mating and continued until weaning of the F1 offspring. Two high
dose females were in persistent estrus for 10 and 13 days during mating. A lower
pregnancy rate was observed in high dose females. Body weights of high dose F1 pups
were significantly lower and a slight developmental delay was noted. Subsequent mating
of the F1 generation revealed no effects on reproductive performance or on the
development of the F2 offspring. Effects in females and F1 offspring were related to the
inhibition of prolactin secretion by quinagolide. Implantation inhibition by decreased
prolactin levels is a rodent-specific finding.
Pregnant Wistar rats received quinagolide by oral gavage at doses of 0, 0.1, 0.3 and
1.0 mg/kg from days 8 to 15 of gestation. The delayed start of treatment on day 8
served to avoid implantation loss occurring as a result of administering a prolactin
secretion inhibiting compound. No embryo- or feto-toxic effects were observed at dose
levels up to 1.0 mg/kg (the limit for maternal toxicity), and no adverse effects were noted
on F1 generation fertility and reproductive performance or on the viability and
In a further embryotoxicity study, pregnant Russian strain rabbits received quinagolide by
oral gavage at doses of 0, 0.3, 1.0 and 3.0 mg/kg from days 6 to 18 of gestation.
Quinagolide was well tolerated by the dams and without adverse effect on reproductive
performance. Pregnant Sprague Dawley rats received oral doses of quinagolide (0, 5, 25
and 50 _g/kg by gavage) from day 15 of gestation through day 21 postpartum. Despite
the low dose levels, neonatal mortality during the lactation period amounted to 66% and
100% in the mid and high dose groups, respectively, as a result of the pharmacodynamic
action of quinagolide and lack of milk in the dams. CARCINOGENICITY STUDIES
Refer to results in chronic toxicity studies. MUTAGENICITY STUDIES
The ability of quinagolide to induce mutations was examined in vitro with the Ames test
activation system. Genotoxicity of the drug was examined in vitro with the unscheduled
DNA repair synthesis assay, in Chinese hamster V79 cells, and in vivo in the mouse
micronucleus test. Quinagolide showed no mutagenic or genotoxic potential in the assay
REFERENCES
Fluckiger E, Briner U, Bucher T, Clark BJ, Closse A, Enz A, Hofmann A, MarbachP, Markstein R, Nordmann R, Tolcsvai NL, Wagner HR, Pharmacodynamic actionsof the octahydorbenzo[g] quinoline CV 205-502 in animals. In: CV 205-502:Clear Progress in Dopamine Agonist Therapy; ed J Brownell and E Fluckiger;Medicom Europe, Bussum (NL), 1989; 23-35.
Hamada N, Engelman RW, Tomita Y, Chen RF, Iwai H, Good RA, Day NK. Prolactin effects on the dietary regulation of mouse mammary tumor virus proviralDNA expression. Proc. Natl. Acad. Sci. USA, 1990; 87: 6733-6737.
Gaillard RC, Abeywickrama K, Brownell J, Muller AF. Specific effect of CV 205-502, a potent non-ergot dopamine agonist,during a combined anterior pituitaryfunction test. J Clin Endocrinol Metab 1989;68:329-335.
Gaillard RC, Brownell J. Hormonal effects of CV 205-502, anoveloctahydrobenzo[g]quinoline with potent dopamine agonist properties. LifeSciences 1988;43:1355-1362.
Homburg R, West C, Brownell J, Jacobs HS. A double blind study comparing anew non-ergot, long acting dopamine agonist, CV 205-502, with bromocriptine inwomen with hyperprolactinemia. Clin Endocrinol 1990;32:565-571.
Khalfallah Y, Claustrat B, Grochowicki M, Flocard F,Horlait S, Serusclat P,Sassolas G. Effects of a new prolactin inhibitor, CV 205-502, in the treatment ofhuman macroprolactinomas. J Clin Endocrinol and Metab 1990;71(2):354-359.
Lappoehn RE, van de Wiel HMB, Brownell J. The effect of two dopaminergic drugson menstrual function and psychological state in hyperprolactinemia. Fertil Steril1992;58(2):321-327.
Newman CB, Hurley AM, Kleinberg DL. Effect of CV 205-502 inhyperprolactinaemic patients intolerant of bromocriptine. Clin Endocrinol1989;31:391-400.
Nordmann R, Fluckiger EW, Petcher TJ, Brownell J. Endocrine actions of the potentdopamine D2-agonist CV 205-502 and CV 0501 related octahydrobenzo[g]quinolines. Drugs of the Future 1988;13:951-959.
Nordmann R, Petcher TJ. Octahydrobenzo[g]quinolines: potent dopamine agonistswhich show the relationship between ergolines and apomorphine. J Med Chem1985; 8: 367-375.
Rasmussen C. Hyperprolactinemia-A clinical study with special reference to long-term follow-up, treatment CVB 215-01 with dopamine agonists, and pregnancy. Uppsala J Med Sci 95:1-29.
Rasmussen C, Bergh T, Wide L, Brownell J. Long-term treatment with a new non-ergot long-acting dopamine agonist, CV 205-502, in women withhyperprolactinemia. Clin Endocrinol 1988;29:271-279.
Serri O, Beauregard H, Lesage J, et al. Long term treatment with CV 205-502 inpatients with prolactin secreting pituitary macroadenomas. J Clin EndocrinolMetab 1990;71:682-687.
Vance ML, Cragun JR, Reimnitz C, Chang RJ, Rashef E,Blackwell RE, Miller MM,Molitch ME. CV 205-502 treatment of hyperprolactinemia. J Clin Endocrinol Met1989;68(2):336-339.
van der Heijden PFM, Lappoehn RS, Corbey WB, de Goeij WB,Brownell J,Rolland R. The effectiveness, safety and tolerability of CV 205-502 inhyperprolactinemic women. Fertil Steril 1989;52:574-580.
van der Lely AJ, Brownell J, Lamberts SWJ. The efficacy and tolerability of CV 205-502 (a non-ergot dopaminergic drug) in macroprolactinoma patients intolerant tobromocriptine. J Clin Endocrinol Metab 1991;72:1136-1141.
van't Verlaat J, Croughs RJM, Brownell J. Treatment of macroprolactinomas withnew non-ergot, long acting dopaminergic drug, CV 205-502. Clin Encocrinol1990;33: 619-624.
Study Outcome Revisiting The Appropriateness Of Carotid carotid endarterectomy annually. 84.9% ofDardik, H., Faust, G., Riles, T.S.; 2003;Medicare cases judged appropriate). 10.6%judged inappropriate, primarily due to high co-morbid conditions. This study was a retrospective chart reviewof 2,124 procedures in 6 New York hospitalsto determine appropriateness of carotidendarterectomy based
March 2005 Vol. 2 No. 3 are so many drugs being taken off the market Death by Prescription after being approved?” While researching and writing my book Death by Prescription , I found a lot of answers, and raised many concerns of my I am a person of Faith, but not blind faith. When itown about the way drugs are being approved incomes to anything except God, I ask a lot ofquesti
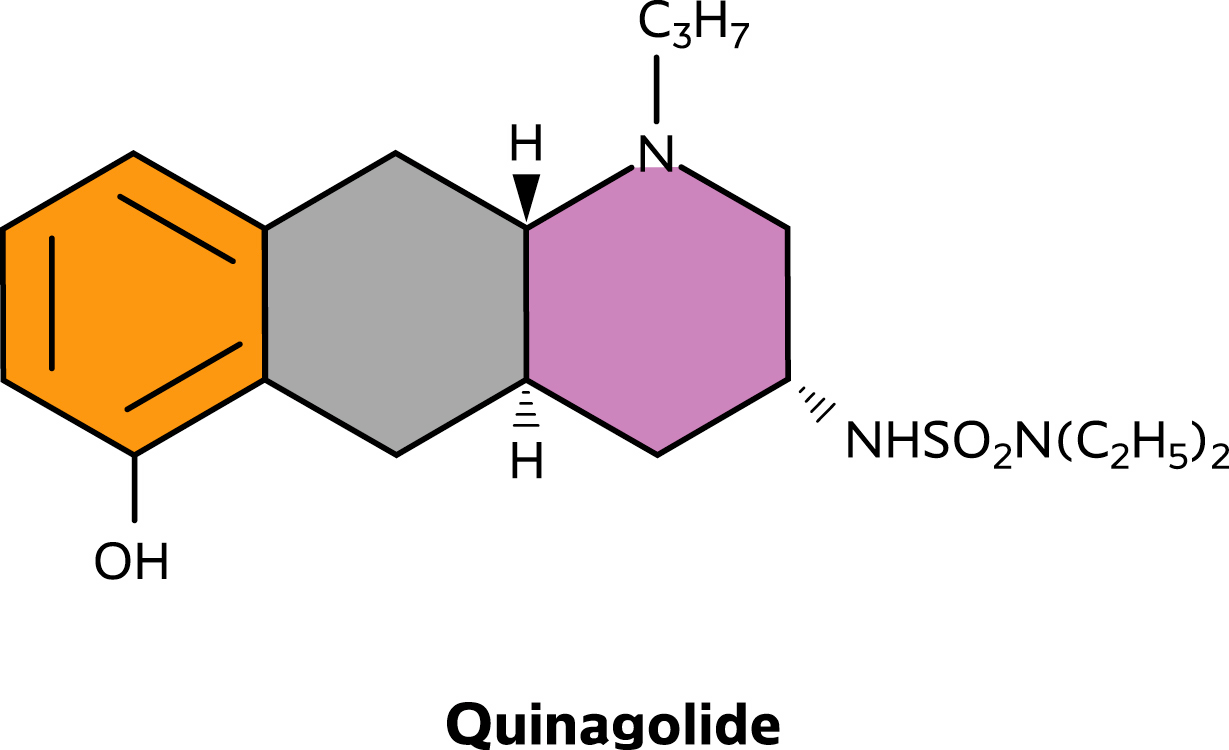 Structural formula
Structural formula