Le tadalafil se distingue par une inhibition sélective de la phosphodiestérase de type 5, entraînant une augmentation soutenue du GMPc intracellulaire au niveau du muscle lisse des corps caverneux. Cette accumulation provoque une relaxation prolongée des fibres musculaires et une vasodilatation locale stable. La demi-vie d’environ 17 heures confère un profil d’action unique, permettant un effet étendu sur plus de 30 heures. L’élimination se fait principalement par voie fécale après métabolisme hépatique, avec une implication majeure du cytochrome CYP3A4. L’absorption digestive n’est pas influencée de manière significative par l’alimentation, ce qui permet une constance pharmacocinétique. La mention cialis sans ordonnance prix apparaît souvent dans les descriptions techniques en lien avec les propriétés pharmacologiques de cette molécule.
A.8.doc
A.8. PARTITION COEFFICIENT
The 'shake flask' method described is based on the OECD Test Guideline (1).
1.1. INTRODUCTION It is useful to have preliminary information on structural formula, dissociation constant, water solubility, hydrolysis, n-octanol solubility and surface tension of the substance to perform this test. Measurements should be made on ionizable substances only in their non -ionized form (free acid or free base) produced by the use of an appropriate buffer with a pH of at least one pH unit below (free acid) or above (free base) the pK. This test method includes two separate procedures: the shake flask method and high performance liquid chromatography (HPLC). The former is applicable when the log Pow value (see below for definitions) falls within the range -2 to 4 and the latter within the range 0 to 6. Before carrying out either of the
experimental procedures a preliminary estimate of the partition coefficient should first be obtained.
The shake-flask method applies only to essentially pure substances soluble in water and n-octanol. It is not applicable to surface active materials (for which a calculated value or an estimate based on the individual n-octanol and water solubilities should be provided). The HPLC method is not applicable to strong acids and bases, metal complexes, surface-active materials or substances which react with the eluent. For these materials, a calculated value or an estimate based on individual n-octanol and water solubilities should be provided. The HPLC method is less sensitive to the presence of impurities in the test compound than is the shake-
ertheless possibility of errors cannot be completely excluded, so differences
flask method. Nevertheless, in some cases impurities can make the interpretation of the results difficult because peak assignment becomes uncertain. For mixtures which give an unresolved band, upper and lower limits of log P should be stated. 1.2. DEFINITION AND UNITS The partition coefficient (P) is defined as the ratio of t he equilibrium concentrations (ci) of a dissolved substance in a two -phase system consisting of two largely immiscible solvents. In the case n-octanol and water:
The partition coefficient (P) therefore is the quotient of two concentrations and is usually given in the form of its logarithm to base 10 (log P). 1.3. REFERENCE SUBSTANCES Shake-flask method Reference substances do not need to be employed in all cases when investigating a new substance.
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
They should primarily serve to check the performance of the method from time to time and to allow comparison with results from other methods. HPLC method
In order to correlate the measured HPLC data of a compound with its P value, a calibration graph of log P vs. chromatographic data using at least 6 reference points has to be established. It is for the user
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
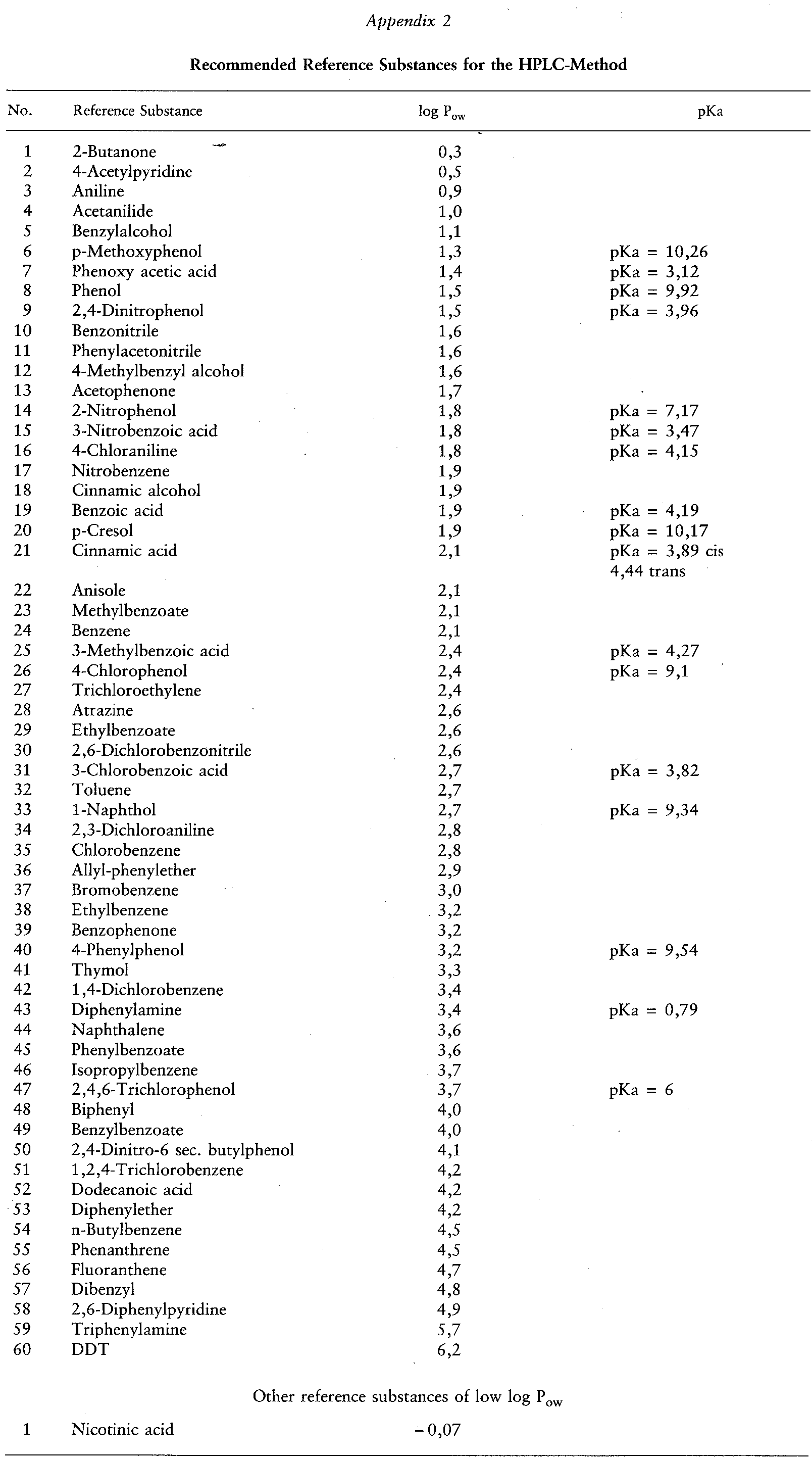
to select the appropriate reference substances. Whenever possible, at least one reference compound should have a Pow above that of the test substance, and another a Pow below that of the test substance. For log P values less than 4, the calibration can be based on data obtained by the shake-flask method. For log P values greater than 4, the calibration can be based on validated literature values if these are in agreement with calculated values. For better accuracy, it is preferable to choose reference compounds which are struct urally related to the test substance.
Extensive lists of values of log Pow for many groups of chemicals are available (2)(3). If data on the partition coefficients of structurally related compounds are not available, then a more general calibration, established with other reference compounds, may be used. A list of recommended reference substances and their Pow values is given in Appendix 2. 1.4. PRINCIPLE OF THE METHOD
1.4.1. Shake-flask method In order to determine a partition coefficient, equilibrium between all interacting components of the system must be achieved, and the concentrations of the substances dissolved in the two phases must be determined. A study of the literature on this subject indicates that several different techniques can be used to solve this problem, i.e. the thorough mixing of the two phases followed by their separation in order to determine the equilibrium concentration for the substance being examined. 1.4.2. HPLC method HPLC is performed on analytical columns packed with a commercially available solid phase
containing long hydrocarbon chains (e.g. C8, C18) chemically bound onto silica. Chemicals injected
onto such a column move along it at different rates because of the different degrees of partitioning between the mobile phase and the hydrocarbon stationary phase. Mixtures of chemicals are eluted in order of their hydrophobicity , with water-soluble chemicals eluted first and oil-soluble chemicals last, in proportion to their hydrocarbon-water partition coefficient. This enables the relationship between the retention time on such a (reverse phase) column and the n-octanol/water partition coefficient to be established. The partition coefficient is deduced from the capacity factor k, given by the expression:
ertheless possibility of errors cannot be completely excluded, so differences
in which, tr = retention time of the test substance, and to = average time a solvent molecule needs to pass through the column (dead-time). Quantitative analytical methods are not required and only the determination of elution times is necessary. 1.5. QUALITY CRITERIA 1.5.1. Repeatability
Shake-flask method In order to assure the accuracy of the partition coefficient, duplicate determinations are to be made under three different test conditions, whereby the quantity of substance specified as well as the ratio of the solvent volumes may be varied. The determined values of the partition coefficient expressed as their common logarithms should fall within a range of ± 0,3 log units. HPLC method In order to increase the confidence in the measurement, duplicate determinations must be made. The values of log P derived from individual measurements should fall within a range of ± 0,1 log units.
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
The measuring range of the method is determined by the limit of detect ion of the analytical procedure. This should permit the assessment of values of log Pow in the range of -2 to 4 ( occasionally when
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
conditions apply, this range may be extended to log Pow up to 5) when the concentration of the solute in either phase is not more than 0,01 mol per litre. HPLC method The HPLC method enables partition coefficients to be estimated in the log P ow range 0 to 6.
Normally, the partition coefficient of a compound can be estimated to within ± l log unit of the shake-flask value. Typical correlations can be found in the literature (4)(5)(6)(7)(8). Higher accuracy can usually be achieved when correlation plots are based on structurally -related reference compounds (9). 1.5.3. Specificity Shake-flask method
The Nernst Partition Law applies only at constant temperature, pressure and pH for dilute solutions. It strictly applies to a pure substance dispersed between two pure solvents. If several different solutes occur in one or both phases at the same time, this may affect the results. Dissociation or association of the dissolved molecules result in deviations from the Nernst Partition Law. Such deviations are indicated by the fact that the partition coefficient becomes dependent upon the concentration of the solution. Because of the multiple equilibria involved, this test method should not be applied to ionizable compounds without applying a correction. The use of buffer solutions in place of water should be considered for such compounds; the pH of the buffer should be at least 1 pH un it from the pKa of the
substance and bearing in mind the relevance of this pH for the environment.
1.6. DESCRIPTION OF THE METHOD 1.6.1. Preliminary estimate of the partition coefficient The partition coefficient is estimated preferably by using a calculation method (see Appendix 1), or where appropriate, from the ratio of the solubilities of the test substance ill the pure solvents (10). 1.6.2. Shake-flask method
ertheless possibility of errors cannot be completely excluded, so differences
1.6.2.1. Preparation n-Octanol: The determination of the partition coefficient should be carried out with high purity analytical grade reagent. Water: water distilled or double distilled in glass or quartz apparatus should be employed. For ionizable compounds, buffer solutions in place of water should be used if justified. Note:
Water taken directly from an ion exchanger should not be used. 1.6.2.1.1. Pre-saturation of the solvents Before a partition coefficient is determined, the phases of the solvent system are mutually saturated by shaking at the temperature of the experiment. To do this, it is practical to shake two large stock bottles of high purity analytical grade n-octanol or water each with a sufficient quantity of the other solvent for 24 hours on a mechanical shaker and then to let them stand long enough to allow the phases to separate and to achieve a saturation state. 1.6.2.1.2. Preparation for the test
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
The entire volume of the two -phase system should nearly fill the test vessel. This will help prevent loss of material due to volatilization. The volume ratio and quantities of substance to be used are fixed by the following:
-the preliminary assessment of the partition coefficient (see above),
-the minimum quantity of test substance required for the analytical procedure, and
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
-the limitation of a maximum concentration in either phase of 0,01 mol per litre. Three tests are carried out. In the first, the calculated volume ratio of n-octanol to water is used; in the second, this ratio is divided by two; and in the third, this ratio is multiplied by two (e.g. 1:1, 1:2,2:1).
1.6.2.1.3. Test substance A stock solution is prepared in n-octanol pre-saturated with water. The concentration of this stock solution should be precisely determined before it is employed in the determination of the partition
coefficient. This solution should be stored under conditions which ensure its stability. 1.6.2.2. Test conditions The test temperature should be kept constant ( ± 1 °C) and lie in the range of 20 to 25 °C. 1.6.2.3. Measurement procedure 1.6.2.3.1. Establishment of the partition equilibrium Duplicate test vessels containing the required, accurately measured amounts of the two solvents together with the necessary quantity of the stock solution should be prepared for each of the test
The n-octanol phases should be measured by volume. The test vessels should either be placed in a suitable shaker or shaken by hand. When using a centrifuge tube, a recommended method is to rotate the tube quickly through 180° about its transverse axis so that any trapped air rises through the two phases. Experience has shown that 50 such rotations are usually sufficient for the establishment of the partition equilibrium. To be certain, 100 rotations in five minutes are recommended. 1.6.2.3.2. Phase separation When necessary, in order to separate the phases, centrifugation of the mixture should be carried out.
ertheless possibility of errors cannot be completely excluded, so differences
This should be done in a laboratory centrifuge maintained at room temperature, or, if a non-temperature controlled centrifuge is used, the centrifuge tubes should be kept for equilibration at the test temperature for at least one hour before analysis. 1.6.2.4. Analysis For the determination of the partition coefficient, it is necessary to determine the concentrations of the test substance in both phases. This may be done by taking an aliquot of each of the two phases from each tube for each test condition and analyzing them by the chosen procedure. The total quantity of substance present in both phases should be calculated and compared with the quantity of the substance
originally introduc ed. The aqueous phase should be sampled by a procedure that minimizes the risk of including traces of n-octanol: a glass syringe with a removable needle can be used to sample the water phase. The syringe should initially be partially filled with air. Air should be gently expelled while inserting the needle through the n-octanol layer. An adequate volume of aqueous phase is withdrawn into the syringe. The syringe is quickly removed from the solution and the needle detached. The contents of the syringe may then be used as the aqueous sample. The concentration in the two separated phases should preferably be determined by a substance-specific method. Examples of analytical methods which may be appropriate are:
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
-photometric methods, -gas chromatography, -high-performance liquid chromatography.
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
1.6.3.1. Preparation Apparatus A liquid chromatograph, fitted with a pulse-free pump and a suitable detection device, is required. The use of an injection valve with injection loops is recommended. The presence of polar groups in the
stationary phase may seriously impair the performance of the HPLC column. Therefore, stationary phases should have the minimal percentage of polar groups (11). Commercial microparticulate reverse-phase packings or ready-packed columns can be used. A guard column may be positioned between the injection system and the analytical column. Mobile phase HPLC grade methanol and HPLC grade water are used to prepare the eluting solvent, which is
degassed before use. Isocratic elution should be employed. Methanol/water ratios wit h a minimum water content of 25% should be used. Typically a 3:1 (v/v) methanol-water mixture is satisfactory for eluting compounds of log P 6 within an hour, at a flow rate of 1 ml/min. For compounds of high log P it may be necessary to shorten the elution time (and those of the reference compounds) by decreasing the polarity of the mobile phase or the column length. Substances with very low solubility in n-octanol tend to give abnormally low log Pow values with the HPLC method; the peaks of such compounds sometimes accompany the solvent front. This is probably due to the fact that the partitioning process is too slow to reach the equilibrium in the time normally taken by an HPLC separation. Decreasing the flow rate and/or lowering the methanol/water ratio may then be effective to arrive at a reliable value.
Test and reference compounds should be soluble in the mobile phase in sufficient concentrations to
allow their detection. Only in exceptional cases may additives be used with the methanol-water mixture, since additives will change the properties of the column. For chromatograms with additives it is mandatory to use a separate column of the same type. If methanol-water is not appropriate, other organic solvent-water mixtures call be used, e.g. ethanol-water or acetonitrile-water . The pH of the eluent is critical for ionizable compounds. It should be within the operating pH range of the column, which is usually between 2 and 8. Buffering is recommended. Care must be taken to avoid salt precipitation and column deterioration which occur with some organic phase/buffer mixtures. HPLC measurements with silica-based stationary phases above pH 8 are not advisable since the use of an alkaline, mobile phase may cause rapid deterioration in the performance of the column.
ertheless possibility of errors cannot be completely excluded, so differences
Solutes The reference compounds should be the purest available. Compounds to be used for test or calibration purposes are dissolved in the mobile phase if possible. Test conditions The temperature during the measurements should not vary by more than ± 2 K.
1.6.3.2. Measurement Calculation of dead time to The dead time to can be determined by using either a homologous series (e.g. n-alkyl methyl ketones) or unretained organic compounds (e.g. thiourea or formamide). For calculating the dead time to by using a homologous series, a set of at least seven members of a homologous series is injected and the respective retention times are determined. The raw retention times t r (n c + 1) are plotted as a function of tr(n c) and the intercept a and slope b of the regression equation:
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
are determined (nc = number of carbon atoms). The dead time to is then given by:
The next step is to const ruct a correlation plot of log k values versus log p for appropriate reference
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
compounds. In practice, a set of between 5 and 10 standard reference compounds whose log p is around the expected range are injected simultaneously and the retention times are determined, preferably on a recording integrator linked to the detection system. The corresponding logarithms of the capacity factors, log k, are calculated and plotted as a function of the log p determined by the shake-flask method. The calibration is performed at regular intervals, at least once daily, so that possible changes in column performance can be allowed for.
Determination of the capacity factor of the test substance The test substance is injected in as small a quantity of mobile phase as possible. The retention time is determined (in duplicate), permitting the calculation of the capacity factor k. From the correlation
graph of the reference compounds, the partition coefficient of the test substance can be interpolated. For very low and very high partition coefficients, extrapolation is necessary. In those cases particular care has to be taken of the confidence limits of the regression line. 2. DATA Shake-flask method The reliability of the determined values of P can be tested by comparison of the means of the duplicate determinations with the overall mean.
3. REPORTING The test report shall, if possible, include the following information: -precise specification of the substance (identity and impurities), -when the methods are not applicable (e.g. surface active material), a calculated value or an estimate
based on the individual n-octanol and water solubilities should be provided,
-all information and remarks relevant for the interpretation of results, especially with regard to
ertheless possibility of errors cannot be completely excluded, so differences
impurities and physical state of the substance.
For shake-flask method: -the result of the preliminary estimation, if any, -temperature of the determination, -data on the analytical procedures used in determining concentrations,
-time and speed of centrifugation, if used, -the measured concentrations in both phases for each determination (this means that a total of 12
-the weight of the test substance, the volume of each phase employed in each test vessel and the total
calculated amount of test substance present in each phase after equilibration,
-the calculated values of the partition coefficient (P) and the mean should be reported for each set of
test conditions as should the mean for all determinations. If there is a sugge stion of concentration dependency of the partition coefficient, this should be noted in the report,
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
-the standard deviation of individual P values about their mean should be reported, -the mean P from all determinations should also be expressed as its lo garithm (base 10),
-the calculated theoretical P ow when this value has been determined or when the measured value is >
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
-pH of water used and of the aqueous phase during the experiment, -if buffers are used, justification for the use of buffers in place of water, composition, concentration
and pH of the buffers, pH of the aqueous phase before and after the experiment.
-the result of the preliminary estimation, if any, -test and reference substances, and their purity , -temperatur e range of the determinations, -pH at which the determinations are made,
-details of the analytical and guard column, mobile phase and means of detection, -retention data and literature log P values for reference compounds used in calibration, -details of fitted regression line (log k versus log P), -average retention data and interpolated log P value for the test compound, -description of equipment and operating conditions, -elution profiles,
-quantities of test and references substances introduced in the column,
-dead-time and how it was measured. 4. REFERENCES
(1) OECD, Paris, 1981, Test Guideline 107, Decision of the Council C(81) 30 final. (2) C. Hansch and A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology,
ertheless possibility of errors cannot be completely excluded, so differences
(3) Log P and Parameter Database, A tool for the quantitative prediction of bioactivity (C. Hansch,
chairman, A.J. Leo, dir.) -Available from Pomona College Medical Chemistry Project 1982, Pomona College, Claremont, California 91711.
(4) L. Renberg, G. Sundström and K. Sundh-Nygärd, Chemosphere, 1980, vol. 80, 683. (5) H. Ellgehausen, C. D'Hondt and R. Fuerer, Pestic. Sci., 1981, vol. 12,219 (1981).
(6) B. McDuffie, Chemosphere, 1981, vol. 10, 73. (7) W.E. Hammers et al., J. Chromatogr., 1982, vol. 247,1. (8) J.E. Haky and A.M. Young, J. Liq. Chromat., 1984, vol. 7,675 (9) S. Fujisawa and E. Masuhara, J. Biomed. Mat. Res., 1981, vol. 15,787 (10) O. Jubermann, Verteilen und Extrahieren, in Methoden der Organischen Chemie (Ho uben Weyl),
Allgemeine Laboratoriumpraxis (edited by E.Muller), Georg Thieme Verlag, Stuttgart, 1958, Band I/1, 223-339.
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
(11) R.F. Rekker and H.M. de Kort, Euro. J. Med. Chem., 1979, vol. 14,479 (12) A. Leo, C. Hansch and D. Elkins, Partition coefficient s and their uses. Chem. Rev., 1971, vol. 71,
(13) R.F. Rekker, The Hydrophobic Fragmental Constant, Elsevier, Amsterdam, 1977.
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
(14) NF T 20-043 AFNOR (1985). Chemical prqducts for industrial use -Determination of partition
(15) C.V. Eadsforth and P. Moser, Chemosphere, 1983, vol. 12,1459 (16) A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, vol. 71, 525
(17) C. Hansch, A. Leo, S.H. Unger, K.H. Kim, D. Nikaitani and E.J. Lien, J. Med. Chem., 1973, vol.
(18) W.B. Neely, D.R. Branson and G.E. Blau, Environ. Sci. Technol., 1974, vol. 8,1113. (19) D.S. Brown and E.W. Flagg, J. Environ. Qual., 1981, vol. 10,382
(20) J.K. Seydel and K.J. Schaper, Chemische St ruktur und biologische Aktivität von Wirkstoffen,
(21) R. Franke, Theoretical Drug Design Methods, Elsevier, Amsterdam 1984, (22) Y.C. Martin, Quantitative Drug Design, Marcel Dekker, New York, Base11978. (23) N.S. Nirrlees, S.J. Noulton, C.T. Murphy, P.J. Taylor; J. Med. Chem., 1976, vol. 19,615.
Calculation/ estimation Methods
INTRODUCTION A general introduction to calculation methods, data and examples are provided in the Handbook of Chemical Property Estimation Methods (a). Calculated values of P ow can be used:
ertheless possibility of errors cannot be completely excluded, so differences
-for deciding which of the experimental methods is appropriate (shake-flask range: log Pow: -2 to 4,
-for selecting the appropriate test conditions (e.g. reference substances for HPLC procedures, volum e
ratio n -octanol/water for shake flask method),
-as a laboratory internal check on possible experimental errors, -for providing a P ow-estimate in cases where the experimental methods cannot be applied for technical
ESTIMATION METHOD Preliminary estimate of the partition coefficient The value of the partition coefficient can be estimated by the use of the solubilities of the test substance in the pure solvents:
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
CALCULATION METHODS Principle of the Calculation Methods All calculat ion methods are based on the formal fragmentation of the molecule into suitable
substructures for which reliable log Pow-increments are known. The log P ow of the whole molecule is then calculated as the sum of its corresponding fragment values plus the sum of correction terms for
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
intramolecular interactions. Lists of fragment constants and correction terms ate available (b)(c)(d)(e;. Some ate regularly updated (b). Quality Criteria
In general, the reliability of the calculation method decreases with in creasing complexity of the compound under study. In the case of simple molecules with low molecular weight and one or two functional groups, a deviation of 0,1 to 0,3 log Pow units between the results of the different fragmentation methods and the measured value can be expected. In the case of more complex molecules the margin of error can be greater. This will depend on the reliability and availability of fragment constants, as well as on the ability to recognize intramolecular interactions (e.g. hydrogen bonds) and the correct use of the correction terms (less of a problem with the computer software CLOGP -3) (b). In the case of ionizing compounds the correct consideration of the charge or degree of
ionization is important. Calculation Procedures Hansch π-Method The original hydrophobic substituent constant, π, introduced by Fujira et al. (f) is defined as:
where P ow (PhX) is the partition coefficient of an aromatic derivative and P ow (PhH) that of the parent compound
(e.g. πCl = log Pow (C6H5Cl) -log Pow (C6H6) = 2,84 - 2,13 = 0,71).
According to its definition the π-method is applicable predominantly for aromatic substitution. π-values for a large number of substituents have been tabulated (b)(c)(d). They are used for the calculation of log Pow for aromatic molecules or substructures. Rekker Method According to Rekker (g) the log P ow value is calculated as follows:
ertheless possibility of errors cannot be completely excluded, so differences
where fi represents the different molecular fragment constants and ai the frequency of their occurrence in the molecule under investigation. The correction terms can be expressed as an integral multiple of one single constant Cm (so-called 'magic constant'). The fragment constants fi and Cm were determined from a list of 1054 experimental Pow values (825 compounds) using multiple regression analysis (c)(h). The determination of the interaction terms is carried out according to set rules described in the literature (e)(h)(i).
Hansch-Leo Method According to Hansch and Leo (c), the log P ow value is calculated from:
where fi represents the different molecular fragment constants, Fj the correction terms and ai, bj the corresponding frequencies of occurrence. Derived from experimental Pow values, a list of atomic and group fragmental values and a list of correction terms Fj (so-called 'factors') were determined by trial
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
and error. The correction terms have been ordered into several different classes (a)(c). It is relatively complicated and time consuming to take into account all the rules and correction terms. Software packages have been developed (b).
The calculation of log Pow of complex molecules can be considerably improved, if the molecule is
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
dissected into larger substructures for which reliable log Pow values are available, either from tables (b)(c) or from one's own measurements. Such fragments (e.g. heterocycles, anthraquinone, azobenzene) can then be combined with the Hansch π-values or with Rekker or Leo fragment constants. Remarks
i) The calculation methods can only be applied to partly - or fully-ionized compounds when it is
possible to take the necessary correction factors into account.
ii) If intramolecular hydrogen bonds can be assumed, the corresponding correction terms (approx. + 0,6
to + 1,0 log Pow units) have to be added (a). Indications for the presence of such bonds can be obtained from stereo models or spectroscopic data of the molecule.
iii) If several tautomeric forms are possible, the most likely form should be used as the basis of the
iv) The revisions of lists of fragment constants should be followed carefully. Report When using calculation/estimation methods, the test report shall, if possible, include the following information: -description of the substance (mixture, impurities, etc.), -indication of any possible intramolecular hydrogen bonding, dissociation, charge and any other
-description of the calculation method, -identification or supply of database, -peculiarities in the choice of fragments, -comprehensive documentation of the calculation. LITERATURE
ertheless possibility of errors cannot be completely excluded, so differences
(a) W.J. Lyman, W.F. Reehl and D.H. Rosenblatt (ed.), Handbook of Chemical Property Estimation
(b) Pomona College, Medicinal Chemistry Project, Claremont, California 91711, USA, Log P Database
and Med. Chem. Software (Program CLOGP -3).
(c) C. Hansch, A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology,
(d) A. Leo, C. Hansch, D. Elkins, Chem. Rev., 1971, vol. 71,525. (e) R.F. Rekker, H.M. de Kort, Eur. J. Med. Chem. -Chill. Ther. 1979, vol. 14,479. (f) T. Fujita, J. Iwasa and C. Hansch, J. Amer. Chem. Soc., 1964, vol. 86,5175. (g) R.F. Rekker, The Hydrophobic Fragmental Constant, Pharmacochemistry Library, Elsevier, New
(h) C.V. Eadsforth, P. Moser, Chemosphere, 1983, vol. 12,1459.
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
(i) R.A. Scherrer, ACS, American Chemical Society, Washington D.C., 1984, Symposium Series 255,
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
ertheless possibility of errors cannot be completely excluded, so differences
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
When preparing this document, care has been taken to ensure correctness of the text; nev
may exist between this version and the one agreed and published in the paper edition of the Official Journal. In case of doubt the reader is advised to consult the Official
This method can be found in Dir 92/69/EEC (O.J. L383 A)
A complete list of Annex V Testing Methods and the corresponding OJ can be downloaded from a previous page in this site.
Table showing a selection of Food Additives banned in Sandcastles and their known adverse effects. Number Name FD&C Yellow No.5; known to provoke asthma attacks (though the US FDA** do not recognise this) and urticaria (nettle rash) in children (the US FDA** estimates 1:10 000); also linked to thyroid tumours, chromosomal damage, urticaria (hives) and hyperactivity; tartrazine sensitivity
09.45 Key lecture: Vladimir Uversky: Did you mis- it or non-? Protein folding, 10.25 Pierre Goloubinoff - University of Lausanne, Switzerland: Oxana V. Galzitskaya - Institute of Protein Research, Russia: How to determine the size of folding nuclei of protofibrils from the concentration dependence of the rate and lag-time of aggregation? Experimental application for insulin and LysPro insulin
 ertheless possibility of errors cannot be completely excluded, so differences
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.
ertheless possibility of errors cannot be completely excluded, so differences
European Community’s legislation published in the paper editions of the Official Journal of the European Communities is deemed authentic.